Introduction
Definition
GLUT4 (Glucose Transporter Type 4, also known as SLC2A4) is an insulin-regulated facilitative glucose transporter protein that mediates glucose uptake into insulin-sensitive tissues, primarily skeletal muscle, cardiac muscle, and adipose (fat) tissue.[1] As a member of the GLUT family of membrane transport proteins, GLUT4 functions as the primary mechanism by which cells respond to insulin signaling by increasing their capacity to absorb glucose from the bloodstream.
Historical Context
GLUT4 was first characterized in the late 1980s through molecular cloning studies that sought to understand insulin-stimulated glucose transport.[2] Researchers discovered that unlike other glucose transporters that reside constitutively at the cell surface, GLUT4 exhibits unique insulin-responsive translocation behavior. This discovery revolutionized our understanding of glucose homeostasis and insulin action, establishing GLUT4 as the molecular link between insulin signaling and cellular glucose uptake. Subsequently, GLUT4 became recognized as a critical player in metabolic diseases, particularly type 2 diabetes mellitus and insulin resistance syndromes.
Scope of Article
This wiki entry provides comprehensive coverage of GLUT4 structure, function, regulation, and clinical significance. The article examines the molecular mechanisms governing GLUT4 translocation, explores its role in glucose homeostasis and metabolic disease, and discusses therapeutic strategies targeting GLUT4 pathways. Furthermore, this entry addresses current research directions and unresolved questions in GLUT4 biology. This article does not cover other members of the GLUT transporter family in detail, focusing specifically on GLUT4’s unique properties and physiological importance.
Detailed Explanation
Core Concept: The GLUT4 Translocation Mechanism
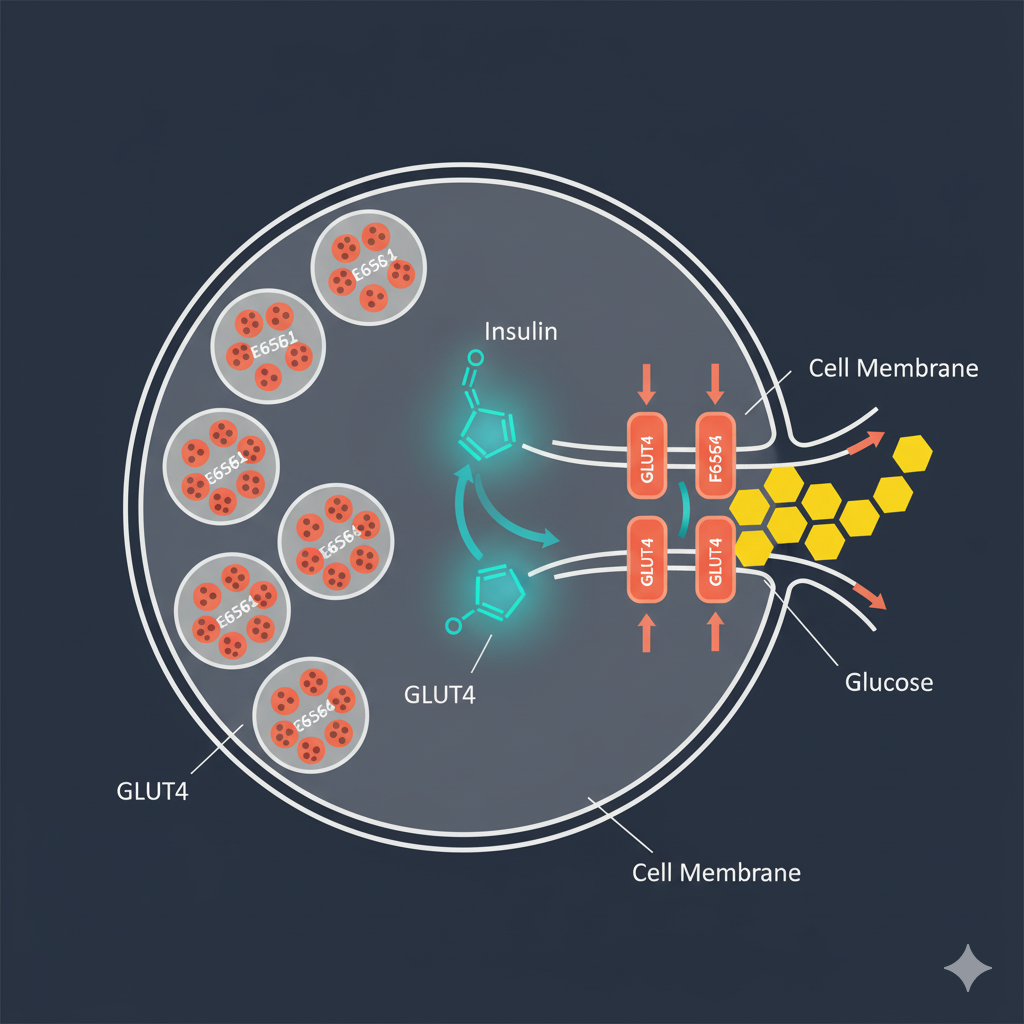
The hallmark feature of GLUT4 biology is its dynamic trafficking between intracellular storage compartments and the plasma membrane in response to insulin and other stimuli.[3] In the basal (non-stimulated) state, approximately 90-95% of cellular GLUT4 resides in specialized intracellular vesicles called GLUT4 storage compartments (GSCs). These vesicles contain high concentrations of GLUT4 protein but remain sequestered away from the cell surface, rendering them functionally inactive for glucose transport.
When insulin binds to its receptor on the cell surface, it initiates a complex signaling cascade involving phosphatidylinositol 3-kinase (PI3K) and the serine/threonine kinase Akt (also known as protein kinase B).[4] This signaling pathway triggers the movement of GLUT4-containing vesicles toward the plasma membrane through a process called exocytosis. The vesicles dock at the membrane, fuse with it, and expose GLUT4 proteins at the cell surface where they can facilitate glucose entry. Consequently, insulin stimulation can increase glucose transport capacity by 10-40 fold depending on the tissue type.
The GLUT4 translocation process is reversible. When insulin levels decline, GLUT4 proteins are retrieved from the plasma membrane through endocytosis and returned to intracellular storage vesicles via recycling endosomes.[5] This dynamic equilibrium between exocytosis and endocytosis allows cells to rapidly adjust their glucose uptake capacity in response to metabolic demands and hormonal signals.
Structural Components and Protein Domains
| Structural Component | Function | Clinical Significance |
|---|---|---|
| 12 Transmembrane Domains | Form the glucose translocation pore; create selective channel for glucose passage | Mutations affecting these domains can impair glucose transport efficiency |
| N-terminal Cytoplasmic Domain | Contains trafficking motifs; regulates vesicle sorting and retention | Critical for insulin-stimulated translocation; target for therapeutic intervention |
| C-terminal Cytoplasmic Domain | Mediates protein-protein interactions; facilitates vesicle docking | Binding site for regulatory proteins; influences translocation kinetics |
| FQQI Motif (Phe-Gln-Gln-Ile) | Endocytosis signal; promotes GLUT4 retrieval from membrane | Defects prevent proper sequestration; lead to constitutive membrane presence |
| Intracellular Loop Regions | Substrate recognition; conformational changes during transport | Determines glucose binding affinity (Km ~5 mM) |
GLUT4 is a 509-amino acid integral membrane protein with a predicted molecular mass of approximately 55 kDa.[6] Its structure includes twelve membrane-spanning α-helices that create a central glucose translocation pore. The protein’s cytoplasmic N- and C-terminal domains contain critical trafficking signals that distinguish GLUT4 from other GLUT family members and enable its unique insulin-responsive behavior.
GLUT4 Isoforms and Tissue Distribution
| Tissue Type | GLUT4 Expression Level | Insulin Response | Physiological Role |
|---|---|---|---|
| Skeletal Muscle | Very High (largest total pool) | 10-20× increase in uptake | Accounts for 70-80% of whole-body glucose disposal |
| Adipose Tissue | High | 20-40× increase in uptake | Glucose storage as triglycerides, metabolic signaling |
| Cardiac Muscle | Moderate-High | 5-10× increase in uptake | Myocardial energy supply, metabolic flexibility |
| Liver | Absent/Minimal | N/A (uses GLUT2) | Relies on constitutive glucose transporters |
| Brain | Absent/Minimal | N/A (uses GLUT1, GLUT3) | Insulin-independent glucose supply required |
Unlike some transporter families that exhibit multiple isoforms, GLUT4 exists as a single gene product (SLC2A4) without known alternatively spliced variants in humans.[7] However, GLUT4 expression levels vary dramatically across different tissues. Skeletal muscle contains the highest total amount of GLUT4 protein and accounts for the majority of whole-body insulin-stimulated glucose disposal due to its large mass. Adipose tissue expresses substantial GLUT4 levels and exhibits the most dramatic fold-increase in glucose transport upon insulin stimulation. Cardiac muscle also expresses GLUT4, which plays important roles in myocardial glucose metabolism and energy supply.
Notably, GLUT4 is absent or expressed at very low levels in most other tissues, including the brain, liver, and pancreatic beta cells, which rely on other glucose transporters for their glucose uptake needs.[8] This tissue-specific expression pattern reflects GLUT4’s specialized role in mediating insulin-dependent glucose disposal in peripheral tissues rather than constitutive glucose supply to all cells.
Scientific Basis
Molecular Biology and Genetics
| Genetic Feature | Details | Functional Significance |
|---|---|---|
| Gene Name | SLC2A4 (Solute Carrier Family 2 Member 4) | Official nomenclature for glucose transporter type 4 |
| Chromosomal Location | Chromosome 17p13 | Genetic mapping for mutation screening |
| Gene Size | ~6 kilobases (kb) | Relatively compact gene structure |
| Number of Exons | 11 exons | Enables alternative splicing regulation (though rare) |
| Transcription Factors | MyoD (muscle), PPARγ (adipose), MEF2, thyroid hormone receptor | Tissue-specific and metabolic regulation of expression |
| Knockout Phenotype (mice) | Homozygous: embryonic lethal; Heterozygous: insulin resistance | Essential for development and glucose homeostasis |
The human GLUT4 gene (SLC2A4) is located on chromosome 17p13 and spans approximately 6 kb of genomic DNA containing 11 exons.[9] Gene expression is regulated by multiple transcription factors that respond to metabolic and hormonal signals. Thyroid hormone, insulin, and exercise training can increase GLUT4 mRNA and protein levels, whereas glucocorticoids and aging tend to reduce GLUT4 expression. Additionally, the muscle-specific transcription factor MyoD and the metabolic regulator PPARγ play important roles in maintaining GLUT4 expression in skeletal muscle and adipose tissue, respectively.
Genetic studies have revealed that complete absence of GLUT4 is embryonically lethal in mice, underscoring its essential physiological role.[10] Heterozygous GLUT4 knockout mice with ~50% reduction in GLUT4 protein develop insulin resistance and glucose intolerance, demonstrating that adequate GLUT4 expression is critical for normal glucose homeostasis. In humans, rare polymorphisms in the SLC2A4 gene have been associated with increased risk of type 2 diabetes, though these associations remain controversial and require further validation.
Biochemical Mechanisms of Glucose Transport
GLUT4 mediates glucose transport through facilitated diffusion, meaning it enables glucose to move down its concentration gradient without requiring ATP hydrolysis.[11] The transporter operates through an alternating access mechanism in which the glucose binding site alternates between facing the extracellular and intracellular environments. When glucose binds to the outward-facing conformation, the protein undergoes a conformational change that exposes the binding site to the cell interior, allowing glucose release into the cytoplasm.
GLUT4 exhibits a Km (Michaelis constant) for glucose of approximately 5 mM, which is close to normal blood glucose concentrations (4-6 mM).[12] This kinetic property means that GLUT4-mediated glucose uptake is highly responsive to changes in blood glucose levels within the physiological range. The transporter shows strong substrate specificity for D-glucose and also transports dehydroascorbic acid (oxidized vitamin C), though glucose remains the primary physiological substrate.
Insulin-Independent GLUT4 Activation
While insulin represents the primary physiological stimulus for GLUT4 translocation, exercise and muscle contraction activate GLUT4 through an entirely insulin-independent mechanism.[13] During muscle contraction, cellular energy depletion activates AMP-activated protein kinase (AMPK), which triggers GLUT4 translocation through signaling pathways distinct from the insulin-PI3K-Akt cascade. This dual regulation provides metabolic flexibility, allowing muscle glucose uptake during exercise even in insulin-resistant states.
Other stimuli can also promote GLUT4 translocation in specific contexts. Hypoxia, certain pharmacological agents like AICAR (an AMPK activator), and some adipokines have been shown to stimulate GLUT4 movement to the plasma membrane.[14] Understanding these alternative activation pathways has therapeutic implications for developing insulin-independent strategies to improve glucose uptake in diabetes.
Clinical and Practical Significance
Role in Type 2 Diabetes and Insulin Resistance
Impaired GLUT4 function represents a central defect in type 2 diabetes mellitus and insulin resistance.[15] Multiple mechanisms contribute to GLUT4 dysfunction in these conditions. Reduced GLUT4 protein expression has been documented in skeletal muscle and adipose tissue of people with type 2 diabetes, with decreases ranging from 20-50% depending on the study and tissue examined. Beyond reduced expression, insulin-stimulated GLUT4 translocation is markedly impaired, meaning even the available GLUT4 protein fails to reach the cell surface appropriately in response to insulin.
The molecular basis of impaired GLUT4 translocation in diabetes involves multiple factors.[16] Chronic hyperglycemia and lipotoxicity induce cellular stress that interferes with insulin signaling pathways. Inflammatory cytokines produced by adipose tissue activate stress kinases that phosphorylate insulin signaling proteins at inhibitory sites, blocking the PI3K-Akt cascade required for GLUT4 translocation. Furthermore, defects in the GLUT4 vesicle trafficking machinery itself have been identified, including alterations in the expression or function of proteins that regulate vesicle docking and fusion at the plasma membrane.
| Metabolic Condition | GLUT4 Expression | GLUT4 Translocation | Glucose Uptake |
|---|---|---|---|
| Normal/Healthy | 100% (baseline) | Normal (10-40× increase with insulin) | Efficient |
| Prediabetes/Early Insulin Resistance | 85-90% | Moderately impaired (5-15× increase) | Reduced |
| Type 2 Diabetes (Established) | 50-80% | Severely impaired (2-5× increase) | Markedly impaired |
| After Exercise Training (8-12 weeks) | 110-130% | Improved/Normalized | Enhanced |
Therapeutic Targeting of GLUT4
| Therapeutic Strategy | Mechanism of Action | Effect on GLUT4 | Clinical Evidence |
|---|---|---|---|
| Metformin | AMPK activation | Enhanced translocation without affecting expression | First-line T2D medication; 25-30% HbA1c reduction |
| Thiazolidinediones (TZDs) | PPARγ agonism | Increased expression (40-60%) and improved translocation | Powerful insulin sensitizers; cardiovascular concerns limit use |
| Aerobic Exercise Training | Chronic AMPK activation, transcriptional upregulation | Increased expression (40-100%) depending on intensity | Comparable to medication effects; 150 min/week recommended |
| Resistance Training | Muscle hypertrophy, increased total GLUT4 pool | Increased expression + muscle mass expansion | Synergistic with aerobic exercise; 2-3 sessions/week optimal |
| Weight Loss/Caloric Restriction | Reduced lipotoxicity, improved insulin signaling | Improved translocation response to insulin | 5-10% weight loss significantly improves insulin sensitivity |
| Emerging: GLUT4 Vesicle Trafficking Enhancers | Target specific trafficking proteins (Rab, SNARE proteins) | Direct enhancement of translocation machinery | Preclinical stage; promising in animal models |
Enhancing GLUT4 expression and/or translocation represents an attractive therapeutic strategy for improving glucose control in diabetes.[17] Several classes of diabetes medications work at least partially through GLUT4-related mechanisms. Thiazolidinediones (TZDs), which are PPARγ agonists, increase GLUT4 gene expression in adipose tissue and improve insulin-stimulated GLUT4 translocation. Metformin enhances GLUT4 translocation through AMPK activation, mimicking some effects of exercise. Additionally, emerging drug candidates targeting specific components of the GLUT4 trafficking machinery are under investigation.
Lifestyle interventions profoundly impact GLUT4 biology.[18] Aerobic exercise training increases GLUT4 protein expression in skeletal muscle by 40-100% depending on training intensity and duration. Resistance training similarly upregulates GLUT4 while also increasing muscle mass, thereby expanding total glucose disposal capacity. Weight loss in obese individuals improves insulin-stimulated GLUT4 translocation even before significant changes in body weight occur, suggesting that reducing lipotoxicity rapidly benefits GLUT4 function.
GLUT4 in Other Metabolic Conditions
Beyond type 2 diabetes, GLUT4 dysfunction contributes to metabolic complications in various conditions. Polycystic ovary syndrome (PCOS), which affects up to 10% of reproductive-age women, features insulin resistance with documented GLUT4 defects in skeletal muscle.[19] Gestational diabetes involves impaired GLUT4 translocation in placental and maternal tissues. Aging is associated with progressive decline in GLUT4 expression and insulin-stimulated glucose transport, contributing to age-related glucose intolerance. Prolonged bed rest and physical inactivity rapidly reduce GLUT4 protein levels, highlighting the importance of regular muscle contraction for maintaining glucose transporter expression.
Controversies and Limitations
Debates in the Field
Several aspects of GLUT4 biology remain contentious among researchers. One ongoing debate concerns whether reduced GLUT4 expression or impaired translocation represents the primary defect in type 2 diabetes.[20] Some studies emphasize decreased GLUT4 protein levels as the dominant factor, while others argue that translocation defects occur even when GLUT4 expression is normal or only mildly reduced. This distinction has implications for therapeutic strategies—should interventions focus on increasing GLUT4 gene expression or on fixing the trafficking machinery?
Another controversial area involves the precise molecular identity of the GLUT4 storage compartment.[21] Researchers disagree about whether GSCs represent a single, specialized organelle type or a heterogeneous collection of vesicles derived from multiple endosomal compartments. Different laboratories have reported conflicting markers and characteristics for GSCs, making it challenging to develop a unified model of GLUT4 storage and mobilization. Resolving this controversy requires improved imaging techniques and more sophisticated cell biological approaches.
Current Knowledge Gaps
Despite decades of research, significant gaps remain in our understanding of GLUT4 regulation. The complete signaling network connecting insulin receptor activation to GLUT4 vesicle fusion at the plasma membrane involves dozens of proteins, many of which have incompletely defined functions. How different signaling pathways (insulin, AMPK, and others) are integrated to control GLUT4 trafficking remains unclear. Additionally, the mechanisms by which chronic insulin resistance becomes established—transitioning from a reversible functional defect to a more permanent reduction in GLUT4 expression—require further investigation.
Another limitation concerns our ability to measure GLUT4 translocation in humans in vivo. Most mechanistic studies have been performed in cell culture or animal models, with limited translation to human physiology.[22] Developing non-invasive imaging methods to quantify GLUT4 translocation in human tissues would represent a major advance, enabling better assessment of therapeutic interventions and individual patient responses.
References
- James DE, Stöckli J, Birnbaum MJ. The aetiology and molecular landscape of insulin resistance. Nat Rev Mol Cell Biol. 2021;22(11):751-771. doi:10.1038/s41580-021-00390-6 PMID: 34285405
- Birnbaum MJ. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell. 1989;57(2):305-315. doi:10.1016/0092-8674(89)90968-9 PMID: 2649253
- Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13(6):383-396. doi:10.1038/nrm3351 PMID: 22617471
- Stöckli J, Fazakerley DJ, James DE. GLUT4 exocytosis. J Cell Sci. 2011;124(Pt 24):4147-4159. doi:10.1242/jcs.097063 PMID: 22247191
- Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5(4):237-252. doi:10.1016/j.cmet.2007.03.006 PMID: 17403369
- Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2-3):121-138. doi:10.1016/j.mam.2012.07.001 PMID: 23506862
- Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298(2):E141-E145. doi:10.1152/ajpendo.00712.2009 PMID: 20009031
- Shepherd PR, Kahn BB. Glucose transporters and insulin action–implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341(4):248-257. doi:10.1056/NEJM199907223410406 PMID: 10413738
- Olson AL, Pessin JE. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr. 1996;16:235-256. doi:10.1146/annurev.nu.16.070196.001315 PMID: 8839927
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15-25. doi:10.1016/j.cmet.2004.12.003 PMID: 16054041
- Gould GW, Holman GD. The glucose transporter family: structure, function and tissue-specific expression. Biochem J. 1993;295(Pt 2):329-341. doi:10.1042/bj2950329 PMID: 8240230
- Uldry M, Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch. 2004;447(5):480-489. doi:10.1007/s00424-003-1085-0 PMID: 12750891
- Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev. 2013;93(3):993-1017. doi:10.1152/physrev.00038.2012 PMID: 23899560
- Zisman A, Peroni OD, Abel ED, et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6(8):924-928. doi:10.1038/78693 PMID: 10932232
- Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101(11):2377-2386. doi:10.1172/JCI1557 PMID: 9616209
- DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32 Suppl 2(Suppl 2):S157-S163. doi:10.2337/dc09-S302 PMID: 19875544
- Klip A, McGraw TE, James DE. Thirty sweet years of GLUT4. J Biol Chem. 2019;294(30):11369-11381. doi:10.1074/jbc.REV119.008351 PMID: 31171647
- Goodyear LJ, Kahn BB. Exercise, glucose transport, and insulin sensitivity. Annu Rev Med. 1998;49:235-261. doi:10.1146/annurev.med.49.1.235 PMID: 9509261
- Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18(6):774-800. doi:10.1210/edrv.18.6.0318 PMID: 9408743
- Abel ED, Peroni O, Kim JK, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409(6821):729-733. doi:10.1038/35055575 PMID: 11217863
- Sadacca LA, Bruno J, Wen J, Xiong W, McGraw TE. Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Mol Biol Cell. 2013;24(16):2544-2557. doi:10.1091/mbc.E13-02-0103 PMID: 23783033
- Sylow L, Kleinert M, Richter EA, Jensen TE. Exercise-stimulated glucose uptake – regulation and implications for glycaemic control. Nat Rev Endocrinol. 2017;13(3):133-148. doi:10.1038/nrendo.2016.162 PMID: 27739515
Related Wiki Entries
- Insulin Signaling Pathway – Molecular cascade connecting insulin receptor activation to cellular responses including GLUT4 translocation
- AMPK (AMP-Activated Protein Kinase) – Energy-sensing enzyme that triggers GLUT4 translocation during exercise independent of insulin
- Type 2 Diabetes Mellitus – Metabolic disease characterized by insulin resistance and impaired GLUT4 function
- Insulin Resistance – Reduced cellular response to insulin affecting GLUT4 translocation and glucose uptake
- Glucose Metabolism – Biochemical pathways for glucose utilization initiated by GLUT4-mediated cellular uptake
External Resources
- NCBI Gene Database – SLC2A4 (GLUT4): Official gene information, genomic location, and expression data
- UniProt – GLUT4 Protein Entry: Comprehensive protein sequence, structure, and functional annotation
- American Diabetes Association: Clinical resources on diabetes management and glucose regulation
- National Institute of Diabetes and Digestive and Kidney Diseases: Research updates and patient information on diabetes and glucose metabolism
Leave a Reply